RNAs That Behave Like Prions
by
Robert Gorter, MD, PhD. et al.
April 10th, 2021
Messenger RNA Definition
Messenger ribonucleic acids (mRNAs) transfer the information from DNA to the cell machinery that makes proteins. Tightly packed into every cell nucleus, which measures just 10 microns in diameter, is a three-meter long double-stranded DNA “instruction manual” on how to build and maintain a human body. In order for each cell to maintain its structure and perform all of its functions, it must continuously manufacture cell-type-specific parts (proteins). Inside each nucleus, a multi-subunit protein called RNA polymerase II (RNAP II) reads DNA and simultaneously fabricates a “message” or transcript, which is called messenger RNA (mRNA), in a process called transcription. Molecules of mRNA are composed of relatively short, single strands of molecules made up of adenine, cytosine, guanine, and uracil bases held together by a sugar-phosphate backbone. When RNA polymerase finishes reading a section of the DNA, the pre-mRNA copy is processed to form mature mRNA and then transferred out of the cell nucleus. Ribosomes read the mRNA and translate the message into functional proteins in a process called translation. Depending on the newly synthesized protein’s structure and function, it will be further modified by the cell, exported to the extracellular space, or will remain inside the cell. The diagram below shows transcription (DNA->RNA) taking place in the cell nucleus where RNAP is RNA polymerase II enzyme synthesizing RNA.
Functions of mRNA
The primary function of mRNA is to act as an intermediary between the genetic information in DNA and the amino acid sequence of proteins. mRNA contains codons that are complementary to the sequence of nucleotides on the template DNA and direct the formation of amino acids through the action of ribosomes and tRNA. mRNA also contains multiple regulatory regions that can determine the timing and rate of translation. In addition, it ensures that translation proceeds in an orderly fashion because it contains sites for the docking of ribosomes, tRNA as well as various helper proteins.
Proteins produced by the cells play a variety of roles, either as enzymes, structural molecules, or as transport machinery for various cellular components. Some cells are also specialized for secreting proteins, such as the glands that produce digestive enzymes or hormones which influence the metabolism of the entire organism.
How do mRNA vaccinations work?
In vaccinations with mRNA, the idea is to have the synthesized mRNA initiate the production of COVID-19 spike proteins being in ribosomes and through this pathway, the host cell would infect itself with self-made spike protein. Natural mRNA carries information from the DNA in the nucleus to the ribosomes as a messenger.
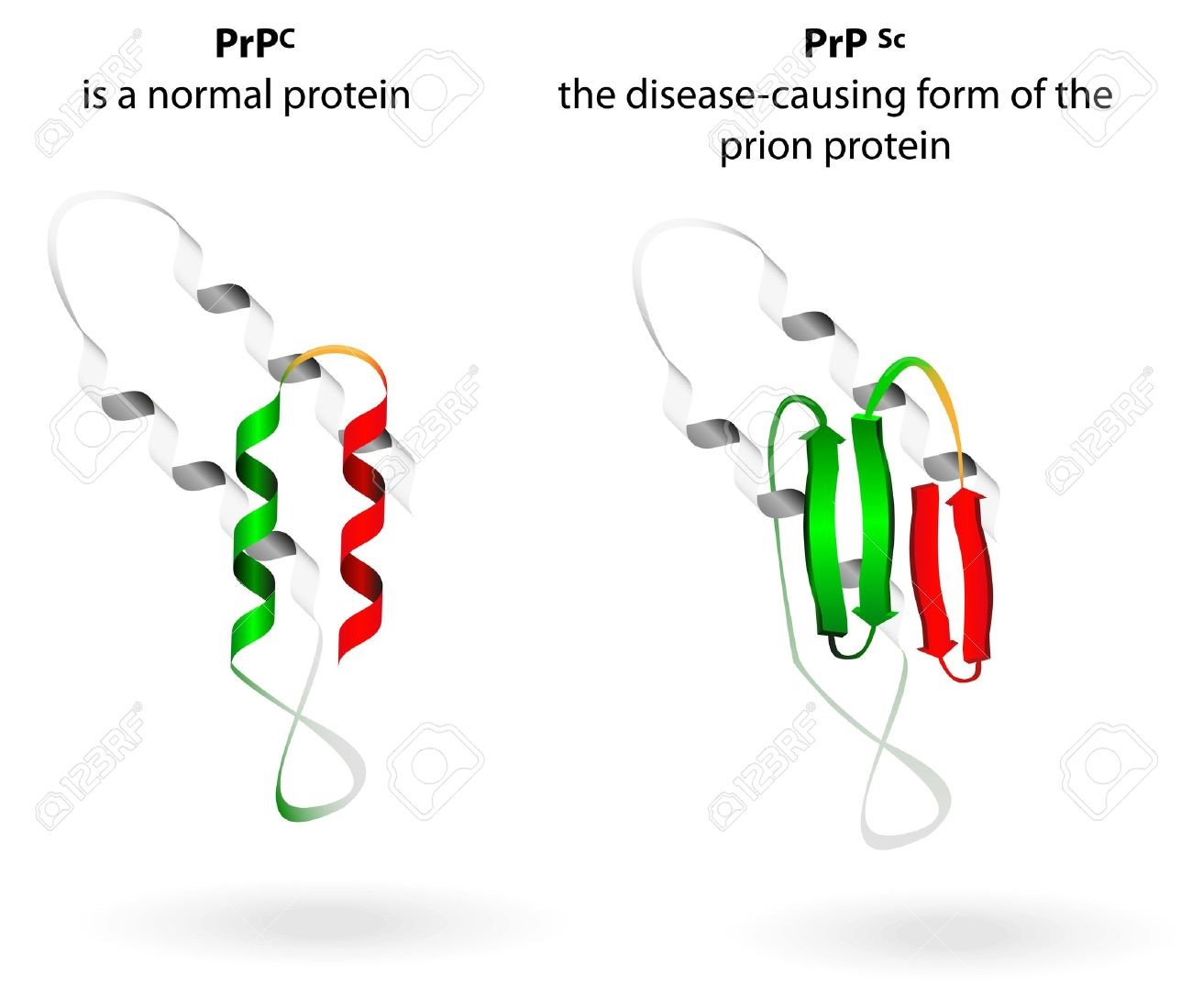
What Are Prions?
Prions are distorted versions of normal proteins found in human and animal brains and other tissues. These distorted (‘misfolded’) proteins damage brain cells, leading to fatal dementias akin to human Alzheimer’s and Parkinson’s diseases. One of the amazing things about prions is that, although they are composed only of proteins and contain no genes more related to RNA-like molecules. They have acquired the ability to be transmitted like any other infectious agents such as viruses or bacteria. Best known prion diseases are mad cow disease of cattle and humans, Creutzfeld-Jakob disease of humans, chronic wasting disease of deer and elk, and scrapie of sheep. A better understanding of prions is important in controlling the threat they represent to animals and humans and for what we can learn about the cause and treatment of the major dementia diseases of humans.
Humans are usually infected by eating infected raw meat (“Beefsteak Tartare” and undercooked meat products) and prions can be found in blood and can be transmitted by blood transfusions and blood products.
Current injections with mRNA contain approx. 70% purified mRNA to inform ribosomes to ensemble COVID-19 spike proteins. What the other 30% mRNA molecules will cause ribosomes to create is unknown but likely aberrant proteins, like prions.
Ribosomes are enzymes composed of many proteins that catalyze the synthesis of proteins from mRNA in the process of translation. Ribosomes exist freely in the cell cytoplasm or remain attached to the endoplasmic reticulum.
Abstract
The term “prion” was originally coined to describe the proteinaceous infectious agents involved in mammalian neurological disorders. More recently, a prion has been defined as a nonchromosomal, protein-based genetic element that is capable of converting the copies of its own benign variant into the prion form, with the new phenotypic effects that can be transmitted through the cytoplasm. Some prions are toxic to the cell, are able to aggregate and/or form amyloid structures, and maybe infectious in the wild, but none of those traits are seen as an integral property of all prions. We propose that the definition of prion should be expanded, to include the inducible transmissible entities undergoing autocatalytic conversion and consisting of RNA rather than protein. We show that when seen in this framework, some naturally occurring RNAs, including ribozymes, riboswitches, viroids, viroid-like retroelements, and PIWI-interacting RNAs (piRNAs), possess several of the characteristic properties of prions.
Well-Informed Opinion/Hypothesis
Mammalian prions: a classical definition. The studies of a peculiar class of infectious diseases in mammals, i.e., transmissible spongiform encephalopathies (TSEs), and the hunt for the elusive agent that causes these diseases, resulted in the notion of an infectious protein, or “prion.” The known prion-caused TSEs include kuru, Creutzfeldt-Jakob disease, Gerstmann-Straussler-Scheinker syndrome, and fatal familial insomnia in humans, as well as scrapie, bovine spongiform encephalopathy, chronic wasting disease, and several other related diseases in mammals. The early evidence in favor of the protein-only agent for scrapie was based on the observations that the infectious subcellular fraction was highly resistant to UV irradiation, unlike the nucleic acids (summarized in reference 1), that the protocol for purification of the infectious moiety required the conditions used to purify certain proteins rather than RNA, and that the estimated molecular weight and other properties of the main component of such a fraction were close to that of a modest-size protein rather than, for example, a virus nucleic acid (2, 3). This was backed up with experiments designed to exclude a role of small infectious RNAs, in particular viroids, in the etiology of scrapie (4, 5). As early as 1967, John S. Griffith outlined several possible mechanisms by which a protein could become inherited as a nonchromosomal genetic element. One of those hypothetical mechanisms stated that a prion is a modified form of a cognate cellular protein, which may bind to the normal form of the same protein (in the simplest case, forming a heterodimer of one prion and one normal copy of the protein) and then turn the normal form into another copy of the prion (6). This predicted mechanism was borne out by the evidence and is at the core of the current definition of any prion.
Scrapie is a fatal, degenerative disease affecting the nervous systems of sheep and goats. It is one of several transmissible spongiform encephalopathies (TSEs), and as such, it is thought to be caused by a prion. Scrapie has been known since at least 1732 and does not appear to be transmissible to humans.
The name scrapie is derived from one of the clinical signs of the condition, wherein affected animals will compulsively scrape off their fleeces against rocks, trees, or fences. The disease apparently causes an itching sensation in the animals. Other clinical signs include excessive lip-smacking, altered gaits, and convulsive collapse.
Scrapie is infectious and transmissible among conspecifics, so one of the most common ways to contain it (since it is incurable) is to quarantine and kill those affected. However, scrapie tends to persist in flocks and can also arise apparently spontaneously in flocks that have not previously had cases of the disease. The mechanism of transmission between animals and other aspects of the biology of the disease are only poorly understood and are active areas of research. Recent studies suggest prions may be spread through urine and persist in the environment for decades. The potential for transmission at birth and from contact with placental tissues is apparent.
In mice, which are susceptible to the sheep scrapie disease, the Sinc gene controlling the incubation period of the disease was identified in 1968 (7). In 1982, it was shown that Sinc encodes protein PrP, which copurifies with the prion fraction (2). Animals lacking the PrP-encoding gene are generally not susceptible to the TSE agents, have a normal life span, do not show abnormalities in the neural system, and do not propagate the prion. In contrast, when animals that encode the wild-type PrP are infected with the TSE agent, they accumulate protease-resistant aggregates of PrP in neural tissues. These aggregates consist of the prion form of PrP, designated PrPSc, which has a high percentage of residues within β-sheets and a large protease-resistant core compared to the normal soluble form PrPC. The β-sheet-rich amyloid aggregates of PrPSc can be obtained in vitro from purified recombinant derivatives of PrPC. Indeed, a line of mice has been developed that, when injected with these aggregates, shows the signs of the disease, and their brain extracts are infectious to many mice lines (8), finally concluding Koch’s triad (see reference 9).
Thus, the canonical definition of prion may include the following: a prion is a protein that is encoded by the cell but is either benign or possibly useful to the organism, hence the preservation of its gene in the genome. Infrequently, it may be transformed into a disease-causing or toxic prion form; the complete set of factors that cause such a transformation is not known but may include physiological stress. Prion causes the conversion of other, benign forms of the same protein into the prion form, i.e., it propagates within the organism, and it is also transmissible to other organisms or sometimes to a different species. The known mammalian prions from amyloids and cause neural phenotypes (Table 1). Notably, the whole infectious cycle does not require any template-directed amplification of nucleic acid, other than the synthesis of the mRNA that is translated into the benign form of a protein.
Criteria and definitions for prions
A broadened definition: prions in fungi. Many extrachromosomal genetic elements, including nucleic acid-based viruses and plasmids, are known in the budding yeast Saccharomyces cerevisiae. Another subset of yeast cytoplasmic inherited factors has been shown to resemble prions in several ways. In one of the first examples, the extrachromosomal element [URE3], which manifests itself by interfering with nitrogen catabolite repression, cannot propagate in the yeast mutant strains lacking the chromosomal copy of the URE2 gene (10). The product of URE2, a glutathione peroxidase-like protein, Ure2p, is able to bind to two transcription factors and thereby repress the genes encoding the subsystem for the utilization of suboptimal nitrogen sources whenever nitrogen is in abundant supply. The phenotype of the ure2 mutants, i.e., the derepression of the systems for utilization of poor nitrogen sources, is the same as the phenotype of [URE3], even though there is no [URE3] in ure2 cells. This led Reed B. Wickner to the idea that [URE3] is a prion of the Ure2p protein—the form of Ure2p in which the “normal” repressor function of the protein is disabled, but the ability of [URE3] to convert Ure2p into extra copies of [URE3] is activated (11). Indeed, [URE3] has been shown to consist of the conformationally altered Ure2p, which can convert “normal” copies of Ure2p into more of [URE3] and form amyloid (12, 13).
In the last 25 years, many other proteins with prion-like behavior have been identified in yeast and other fungi; two of the best-studied ones are [PSI] of S. cerevisiae, the prion form of Sup35 protein that is one of the two subunits of the translation termination complex Sup35-Sup45 (14), and [Het-s] of Podospora anserina, the prion form of the HET-s protein involved in heterokaryon incompatibility, a cell death reaction preventing mating of genetically distant strains and likely protecting the cells from infection by exotic viruses and plasmids—an example of a beneficial prion (15). The molecular mechanisms of the prion-like behavior of those proteins in vivo and in vitro have been studied in some detail, and the debates on the possible biological role of prions, most of which seem to be disadvantageous to their hosts, are ongoing (16–21).
With the extension of the concept of prion to the proteins conferring inheritable phenotypes to yeast and other fungi, the prion definition was modified (Table 1 and Fig. 1). The neural diseases are not applicable to yeast, and even the disease/sickness/loss of fitness in the host is not universal in yeast prions—in the expanded definition, these are replaced by a screenable phenotype. Other integral components of the prion definition, however, still hold true for the yeast prions. Specifically, prion proteins are encoded by cellular genes; the benign form of a protein may be converted into a prion conformation either spontaneously and rarely, or with higher frequency by the action of another copy of a prion; and the prion-associated phenotype is transmitted between cells together with the prion form of the protein. In addition, three operational criteria have been proposed that help to identify candidate prions (22): (i) prions in yeast may be revealed by the fact that the rate of their emergence is increased when the cognate gene is overexpressed, (ii) prions in yeast are often reversibly curable, and (iii) prion phenotype mimics the loss-of-function phenotype of the cognate gene (the last, however, is true only if the prion form is the one in which the normal function of a protein is inactivated; if prion is the active form of a protein, this will manifest as a gain of function).
Protein prions and RNAs that may behave like prions
In all panels, the dark blue lines and shapes indicate the benign forms of proteins or RNAs, red lines and shapes indicate the prion forms, solid arrowed lines indicate the direction of the reaction, and broken arrowed lines indicate the autocatalytic cleavages (gray for the relatively inefficient reactions and black for the more efficient ones). (A) A general scheme of protein prion induction and propagation. The benign form of a protein is converted into the prion form only rarely and spontaneously (left), but once formed, it is able to turn more copies of the benign form into the prion form (center), and in many cases to form aggregates in the cell (right). (B) The “Viennese prison.” (C) A putative prion-like derivative of the glmS ribozyme/riboswitch. The GlcN6P-dependent version described in the text is shown. The green letter G indicates the GlcN6P ligand. The engineered ribozyme requires the presence of a ligand for activity but cleaves with reduced efficiency when acting in cis, and with relatively high efficiency when acting in trans. (D) A putative viroid-derived system engineered to possess prion-like properties. The concatemeric plus-strand viroid RNA is transcribed from the integrated DNA copies (black wavy lines). The engineered HHR region within the viroid RNA processes the concatemer into the unit-length viroids with reduced efficiency when acting in cis, and with relatively high efficiency when acting in trans. (E) Prion-like properties of the ping-pong mechanism of piRNA production. The genomic copies of piRNA clusters and evolutionarily related active transposon copies are shown by black wavy lines, and the enzymes from different protein families that process the piRNA precursors into the mature piRNAs are depicted as gemstones of various colors.
The molecular properties of the benign and prion protein forms in fungi are of particular interest in the context of the new definition of a prion. Though many fungal prions contain protein segments that under some circumstances can form specific amyloid-like, proteinase K-resistant arrangements of parallel in-register β-sheets in vivo and in vitro, this has not been shown for all prions. Moreover, yeast prions do not have to form amyloid at all. Such is the case of the vacuolar protease B (PB, encoded by the prb1 gene), which is synthesized as an inactive zymogen precursor that must be activated through the sequential removal of regions at both termini by another protease (protease A/Pep4p) and by a mature copy of PB. In the strains where Pep4p is deleted, mature Prb1p can in rare cases activate its own precursor. This conversion of an inactive to active form can propagate within a cell and pass between cells, satisfying the definition of a prion (23, 24); in this case, the prion, called [β]—no connection to β-sheets in amyloids—is the same as the active form of protease B. In the absence of Pep4p, [β] is required for survival in the stationary phase and for meiotic sporulation. With the admittance of [β] to the class of prions, the definition of a prion must be modified again, to allow that the chemical composition of the prion and nonprion forms do not have to be exactly the same—protein processing may be permitted. This should not be seen as an unprecedented departure from the prion conventions, given that the posttranslational processing events, such as glycosylation, play a role in the expression of the infectious phenotypes in mammalian PrP (25). The case of prion [β] argues for omitting the amyloid formation from the list of essential properties of a prion. Two other recently described yeast prions, [SMAUG+], the product of the Vts1 gene, and [ESI+], the product of the Snt1 gene, also do not appear to form amyloids, though both may form other kinds of aggregates (26, 27).
Can there be RNA prions?
We propose to take another step in expanding the definition of a prion and admit the possibility of the prion-like behavior in another class of biopolymers, namely, RNA. Even though RNA for a long time has been seen primarily as a carrier of genetic information or facilitator of protein synthesis, it is now clear that RNA molecules may perform catalytic and regulatory functions that do not involve encoding proteins but instead rely on the enzymatic activity, ligand-binding ability, or capacity for dynamic structural rearrangements of RNA itself. In the rest of this paper, we outline the definition of an RNA prion and review several classes of RNA that may come close to satisfying this definition.
Most criteria for a protein-based prion can be generalized for RNA as an RNA prion is encoded by the cellular gene but inherited extrachromosomally; it has a phenotype that is due to the function of the RNA itself, not of its encoded protein if such protein exists; and the phenotype mediated by an RNA prion is inducible and transmissible. RNA prions have two forms, a benign and a phenotype-causing one; analogously to the case of the protein prion [β], conversion between the benign and prion form of RNA may involve RNA processing. The benign form may undergo a rare conversion to the prion form, perhaps stimulated by stress or other external factors. When the prion form is already present, the rate of conversion of benign copies to the prion form increases.
Several of the above properties have been observed in some naturally occurring or computationally designed RNA molecules. We next review four cases of the RNAs that may come close to satisfying the expanded prion definition. We note that, as with yeast protein prions, a neurological phenotype is not required for an RNA prion, and neither is amyloid formation.
“Viennese prion”
Stefan Badelt and coworkers have presented the results of a computational design of an RNA satisfying the following condition: the molecule must be bistable, i.e., it must preferentially be in one structural state when it is a monomer and preferentially adopt another structural state when it is in a dimer (28). Thermodynamic calculations have been done to determine the stability of both forms and ensure that they are separated by a ridge on the folding landscape, without any local free energy minima close to either of the two stable conformations. The designed putative RNA prion (the authors modestly called it an “RNA with prion-like properties”) is a circular RNA consisting of 49 nucleotides, which exhibits an extensive pairing in both conformations. One form, called S1, persists as a monomer, while the other, S2, has two stem-loops that are likely to hybridize via a kissing-loop interaction when present on two different molecules, stabilizing the S2 form in a dimer. The two loops are sterically unlikely to interact when they are in the same molecule, but either of the loops can enter a kissing interaction with the complementary sequence on another copy of S1. Such a complementary region is partly occluded by alternative base pairing within S1, but its interaction with S2 melts this alternative pairing, forcing S1 to change its conformation into S2 (Table 1 and Fig. 1). These properties, once realized in a physical RNA molecule synthesized in vitro, will satisfy several criteria of an RNA prion; of course, biological and genetic criteria, such as the phenotype caused by this “Viennese prison,” as well as the conditions of its induction and curing, may only be considered after the introduction of such a construct into a living cell.
Self-processing riboswitches: the glmS example. Riboswitches are structured regions that are found in the non-coding portions of mRNAs in all three domains of life. In bacterial and archaeal mRNAs, riboswitches are more often located in the 5′ untranslated regions (UTRs), and in eukaryotic mRNAs, they are found mostly in the 3′ untranslated regions and in introns. Many riboswitches regulate gene expression, typically by binding small metabolites and inducing changes in the synthesis or stability of the mRNAs in which they are embedded (29, 30). In an elaboration of this theme, a riboswitch from the 5′ UTR of the mRNA of the glmS gene, conserved in many Gram-positive bacteria, was shown to self-cleave in the presence of glucosamine-6-phosphate (GlcN6P). The glmS open reading frame encodes glutamine-fructose-6-phosphate amidotransferase, which is the terminal enzyme of GlcN6P synthesis. Self-cleavage of the 5′ UTR in the glmS mRNA exposes the transcript to degradation, therefore ensuring the shutdown of metabolite production when it has accumulated in the cell (31).
Various derivatives of the game riboswitch-ribozyme have been designed experimentally or selected in vivo evolution experiments, including the forms active in the absence of GlcN6P and a variant efficiently cleaving its own copies in trans (32, 33). It is conceivable that these properties could be combined to generate new RNAs, which may display prion-like behavior. One version of such a putative RNA prion would be active in the presence of GlcN6P; in such a case, one could engineer an embedded copy of the ribozyme that cleaves inefficiently in cis but releases a product that cleaves the embedded copies of itself more efficiently in trans. The RNA prion then would be inducible by GlcN6P, the prion infection would propagate in the cell, and the RNA prions transferred into another cell would initiate prion formation there, as long as GlcN6P is present (Table 1 and Fig. 1). This would provide the same catabolite repression phenotype as the one mediated in cis by the unmodified glmS ribozyme-riboswitch, except for perhaps different response kinetics. In a GlcN6P-independent version of glmS, the released copy of the ribozyme could be engineered to be more active than the embedded one; this would be essentially a toxic prion, similar to the TSE prions and many yeast protein-based prions.
Viroids and viroid-like elements.
By the end of the 1960s, Theodor Diener characterized the first of a new class of plant pathogens constituted by a naked small circular covalently closed RNA molecule (34). Since that seminal discovery, about 45 different viroid species have been described and classified into two taxonomic families according to sequence similarity and functional properties (35). The mature circular form of viroids (which superficially resembles the secondary structure of the S1 form of the “Viennese prion” described above, though viroid genomic RNAs are longer) is infectious and transmissible between cells and organisms. Members of the larger Pospiviroidae family replicate in the nucleus of susceptible cells via a rolling-circle mechanism, relying on the cell for the three enzymatic activities required for the replication cycle: a DNA-dependent RNA polymerase as replicas, an endonuclease to process the oligomeric intermediates into monomeric genomes, and a ligase to close these monomers into mature circular molecules (36). The members of the smaller Avsunviroidae family replicate in the chloroplast and also require the replicase and ligase activities from the cell, but they do not require a cellular endonuclease, as they all encode, in genomic and antigenomic strands, self-cleaving hammerhead ribozymes (HHRs), which process the oligomers into genomic monomers.
The HHR also has been described in other viroid-like molecules, such as the viroid-like satellites of nepoviruses and luteoviruses (also known as virusoids) and human hepatitis δ virus RNA. Interestingly, several tandem cDNA copies of a viroid-like molecule known as Carnation small viroid-like RNA (CarSV) are embedded into the DNA genome of a plant pararetrovirus, Carnation etched ring virus, and, via the integrated copies of this virus, have made their way into the genomes of carnation plants (37–39), satisfying the criterion of a genome-encoded factor. Unlike most bona fide viroids, these viroid-like molecules require the assistance of a helper virus to ensure their reproduction and transmission among hosts.
The HHR is composed of a catalytic core of conserved nucleotides flanked by three helices, two of which are involved in essential tertiary interactions that facilitate the self-catalysis of the phosphodiester backbone (40). In recent years, an increasing number of noncoding circular RNAs, varying in size between 100 and 1,000 nucleotides, have been reported in both plants and animals; all these genetic elements contain self-cleaving functional HHRs and have been called “retrozymes” and suggested to have evolved from Penelope-like retroelements which also harbor HHRs (34, 48).
It has been established long ago that “prions are not viroids,” in the specific sense that there is no small viroid-like RNA associated with the protein fraction that transmits TSE, and that various inactivation experiments with mammalian infectious prions suggest the pattern of sensitivity typical of a protein, not of viroid RNA (4). However, if the definition of prion is extended to genetic elements made of RNA, it becomes evident that some real-life viroids of the Avsunviroidae family and viroid-like molecules containing HHR either display or can be engineered to display several prion-like features.
During the replication cycle of HHR viroids, cleavage turns the nontransmissible RNA multimers of the genomic-strand RNA into the infectious entity, the unit-length circular RNA, thus satisfying the requirement of the self-medicated conversion from the benign to prion form. The wild-type viroid HHRs are inactive when in monomers, but synthetic derivatives of the hammerhead ribozymes that are able to cleave in trans, usually at a reduced rate, have been obtained (41–43, 48).
A system consisting of a genomically integrated concatemer of a viroid-like cDNA, engineered to be expressed in an inducible fashion but, as in the case of CarSV, unable to replicate autonomously, would go a long way toward satisfying an RNA prion definition (Table 1). Such a system, although derived from a viroid, would not require RNA replication for the infectivity and prion-like behavior—the transcription of the gene integrated into the host genome will suffice, just as in the case of the protein prions. To confer even more prion properties to a CarSV-like RNA, its relative rates of cleavage in cis and in trans would have to be reversed, so that a noninfectious multimeric transcript is processed into the unit-length forms in cis only rarely, but the mature viroid would cleave in trans more efficiently (Fig. 1). In such a design, the prion property of the entire system would be maintained by an HHR-containing RNA element that propagates itself by cutting the benign multimeric RNA forms that the cell produces by transcribing the integrated concatemers; the products of the processing are the unit-length viroid-like RNAs. It is likely that the knowledge about viroid-encoded HHRs is already sufficient to engineer such a construct, which could serve as another model of an RNA prion in vivo.
Small piRNAs produced by the ping-pong mechanism.
The PIWI-interacting RNA (piRNA) pathway is a gene silencing system that is thought to protect animal germline cells from the deleterious effects of the activity of transposable elements (TEs). Experiments in the fruit fly have shown that gamete development is dependent on the expression of a specific fraction of small (23- to 30-nucleotide) RNAs, which are complementary to transposable elements and some other genome repeats. Such small RNAs are also found in the reproductive tissues of vertebrate animals, and in all species, they are associated with the members of a particular clade of the Argonaute proteins, the PIWI family, from which the name piRNA is derived (44). Multiple piRNAs are genomically encoded, typically by piRNA clusters, which are the loci of deleted or nested copies of DNA transposons that have lost the ability to transpose. Transcription of either one or both DNA strands in these clusters produces piRNA precursors, which are bound by a cascade of RNA-binding and RNA-compartmentalizing factors and then processed into the mature “primary” piRNAs by at least two RNases. The primary piRNAs associate with three proteins from the PIWI clade, and interestingly, the sequences bound to the fruit fly PIWI-clade proteins Piwi and Aubergine on the one hand and Ago3 protein on the other hand show orientation bias, different terminal sequences, and 10-nucleotide overlap (44, 45). These and other observations led to the idea of the ping-pong model, in which the piRNAs derived from the transcripts from one DNA strand aid the formation of complementary overlapping piRNAs, which are produced from the transcripts encoded by another strand. Since piRNA clusters are derived from the defective TEs, the full-length transcripts from the non-defective TEs are also targeted, and ping-pong amplification simultaneously generates more piRNA and silences the target TEs by inactivating their transcripts (as well as by other mechanisms, reviewed in reference 45, that are not considered here).
If we stretch the RNA prion framework somewhat further than in the three previous examples, the system of piRNA production may reveal certain features in common with prions. In this view, the long precursor transcripts of the primary piRNA loci may be seen as the inactive form of the prion; with RNA processing being permitted as part of the prion lifestyle, the piRNA would be the active form of a prion beneficial to the cell (Table 1 and Fig. 1). Primary piRNAs induce the production of more copies of themselves; the amplified secondary piRNAs can be transmitted between cells and, at least in the nematode Caenorhabditis elegans, between generations (46).
Here, we examined the plausibility of expanding the definition of a prion beyond what has been done before, to include the inducible and transmissible RNA agents that can autocatalytically convert themselves from inactive to the active form. We have shown that several designed or naturally occurring classes of RNAs come close to satisfying such a definition. One might disagree with the appropriation of the term “prion” to something that is not made of protein (note, however, that “prion” as an acronym of “proteinaceous infectious agent” is itself malformed—see discussion in reference 47). Linguistic concerns notwithstanding, we think that adding a new dimension to the concept of prion helps to assess the robustness of the concept itself, as well as its applicability to various phenomena in molecular biology. In addition, the analysis presented above immediately suggests several new modalities with interesting properties, which may be constructed and tested by synthetic biologists.
ACKNOWLEDGMENTS
A.R.M. is a Program Director at the National Science Foundation (NSF), an agency of the U.S. Government; his work was supported by the NSF’s Independent Research/Development and Long-Term Professional Development Programs, but the statements and opinions expressed herein are made in the personal capacity and do not constitute the endorsement by NSF or the government of the United States. S.F.E. was supported by grants BFU2015-65037-P (Spain Agencia Estatal de Investigación-FEDER) and PROMETEOII / 2014/012 (Generalitat Valenciana).
This is a work of the U.S. Government and is not subject to copyright protection in the United States.
Arcady R. Mushegian, Santiago F. Elena, Michael J. Imperiale, (Editor in American Society for Microbiology) / DOI: 10.1128 / mSphere.00520-20
What is the bottom line?
None of the commercially available mRNA injections are pure in the synthetically manufactured mRNA and initiate in the host cell ribosomes pure COVID-19 spike proteins. Most of these 30% unidentified half broken-off mRNA molecules will give rise to the manufacturing of abnormal and 100% foreign protein structures; including prions.
In animals and humans, it takes a few years for prions to develop the clinical disease as they usually cause disease on the basis of chronic, degenerative inflammation,
References
1.↵Alper T. 1972. The nature of the scrapie agent. J Clin Pathol Suppl (R Coll Pathol) 25:154–155. doi:10.1136/jcp.25.Suppl_6.154.FREE Full TextGoogle Scholar
2.↵Bolton DC, McKinley MP, Prusiner SB. 1982. Identification of a protein that purifies with the scrapie prion. Science 218:1309–1311. doi:10.1126/science.6815801.Abstract/FREE Full TextGoogle Scholar
3.↵Diringer H, Gelderblom H, Hilmert H, Özel M, Edelbluth C, Kimberlin RH. 1983. Scrapie infectivity, fibrils, and low molecular weight protein. Nature 306:476–478. doi:10.1038/306476a0.CrossRefPubMedGoogle Scholar
4.↵Diener TO, McKinley MP, Prusiner SB. 1982. Viroids and prions. Proc Natl Acad Sci U S A 79:5220–5224. doi:10.1073/pnas.79.17.5220.Abstract/FREE Full TextGoogle Scholar
5.↵Bellinger-Kawahara C, Diener TO, McKinley MP, Groth DF, Smith DR, Prusiner SB. 1987. Purified scrapie prions resist inactivation by procedures that hydrolyze, modify or shear nucleic acids. Virology 160:271–274. doi:10.1016/0042-6822(87)90072-9.CrossRefPubMedWeb of ScienceGoogle Scholar
6.↵Griffith JS. 1967. Nature of the scrapie agent: self-replication and scrapie. Nature 215:1043–1044. doi:10.1038/2151043a0.CrossRefPubMedWeb of ScienceGoogle Scholar
7.↵Dickinson AG, Meikle VM, Fraser H. 1968. Identification of a gene that controls the incubation period of some strains of scrapie agent in mice. J Comp Pathol 78:293–299. doi:10.1016/0021-9975(68)90005-4.CrossRefPubMedWeb of ScienceGoogle Scholar
8.↵Scott M, Foster D, Mirenda C, Serban D, Coufal F, Wälchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, Westaway D, Prusiner SB. 1989. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59:847–857. doi:10.1016/0092-8674(89)90608-9.CrossRefPubMedWeb of ScienceGoogle Scholar
9.↵Zabel MD, Reid C. 2015. A brief history of prions. Pathog Dis 73:ftv087. doi:10.1093/femspd/ftv087.CrossRefPubMedGoogle Scholar
10.↵Aigle M, Lacroute F. 1975. Genetical aspects of [URE3], a non-mitochondrial, cytoplasmically inherited mutation in yeast. Mol Gen Genet 136:327–335. doi:10.1007/BF00341717.CrossRefPubMedWeb of ScienceGoogle Scholar
11.↵Wickner RB. 1994. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264:566–569. doi:10.1126/science.7909170.Abstract/FREE Full TextGoogle Scholar
12.↵Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. 1999. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 283:1339–1343. doi:10.1126/science.283.5406.1339.Abstract/FREE Full TextGoogle Scholar
13.↵Speransky VV, Taylor KL, Edskes HK, Wickner RB, Steven AC. 2001. Prion filament networks in [Ure3] cells of Saccharomyces cerevisiae. J Cell Biol 153:1327–1336. doi:10.1083/jcb.153.6.1327.Abstract/FREE Full TextGoogle Scholar
14.↵Kushnirov VV, Vishnevskaya AB, Alexandrov IM, Ter-Avanesyan MD. 2007. Prion and nonprion amyloids: a comparison inspired by the yeast Sup35 protein. Prion 1:179–184. doi:10.4161/pri.1.3.4840.CrossRefPubMedWeb of ScienceGoogle Scholar
15.↵Saupe SJ. 2011. The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Semin Cell Dev Biol 22:460–468. doi:10.1016/j.semcdb.2011.02.019.CrossRefPubMedGoogle Scholar
16.↵Wickner RB, Edskes HK, Ross ED, Pierce MM, Baxa U, Brachmann A, Shewmaker F. 2004. Prion genetics: new rules for a new kind of gene. Annu Rev Genet 38:681–707. doi:10.1146/annual.genet.38.072902.092200.CrossRefPubMedWeb of ScienceGoogle Scholar
17.↵Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. 2012. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 482:363–368. doi:10.1038/nature10875.CrossRefPubMedWeb of ScienceGoogle Scholar
18.↵Holmes DL, Lancaster AK, Lindquist S, Halfmann R. 2013. Heritable remodeling of yeast multicellularity by an environmentally responsive prion. Cell 153:153–165. doi:10.1016/j.cell.2013.02.026.CrossRefPubMedWeb of ScienceGoogle Scholar
19.↵Jarosz DF, Brown JCS, Walker GA, Datta MS, Ung WL, Lancaster AK, Rotem A, Chang A, Newby GA, Weitz DA, Bisson LA, Lindquist S. 2014. Cross-kingdom chemical communication drives a heritable, mutually beneficial prion-based transformation of metabolism. Cell 158:1083–1093. doi:10.1016/j.cell.2014.07.025.CrossRefPubMedGoogle Scholar
20.↵Wickner RB, Shewmaker FP, Bateman DA, Edskes HK, Gorkovskiy A, Dayani Y, Bezsonov EE. 2015. Yeast prions: structure, biology, and prion-handling systems. Microbiol Mol Biol Rev 79:1–17. doi:10.1128/MMBR.00041-14.Abstract/FREE Full TextGoogle Scholar
21.↵Manjrekar J. 2017. Epigenetic inheritance, prions and evolution. J Genet 96:445–456. doi:10.1007/s12041-017-0798-3.CrossRefGoogle Scholar
22.↵Wickner RB, Edskes HK, Roberts BT, Baxa U, Pierce MM, Ross ED, Brachmann A. 2004. Prions: proteins as genes and infectious entities. Genes Dev 18:470–485. doi:10.1101/gad.1177104.FREE Full TextGoogle Scholar
23.↵Roberts BT, Wickner RB. 2003. Heritable activity: a prion that propagates by covalent autoactivation. Genes Dev 17:2083–2087. doi:10.1101/gad.1115803.Abstract/FREE Full TextGoogle Scholar
24.↵Roberts BT, Wickner RB. 2004. A new kind of prion: a modified protein necessary for its own modification. Cell Cycle 3:100–103. doi:10.4161/cc.3.2.642.CrossRefPubMedGoogle Scholar
25.↵Cancellotti E, Mahal SP, Somerville R, Diack A, Brown D, Piccardo P, Weissmann C, Manson JC. 2013. Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J 32:756–769. doi:10.1038/emboj.2013.6.Abstract/FREE Full TextGoogle Scholar
26.↵Chakravarty AK, Smejkal T, Itakura AK, Garcia DM, Jarosz DF. 2020. A non-amyloid prion particle that activates a heritable gene expression program. Mol Cell 77:251–265.e9. doi:10.1016/j.molcel.2019.10.028.CrossRefGoogle Scholar
27.↵Harvey ZH, Chakravarty AK, Futia RA, Jarosz DF. 2020. A prion epigenetic switch establishes an active chromatin state. Cell 180:928–‐940.e14. doi:10.1016/j.cell.2020.02.014.CrossRefGoogle Scholar
28.↵Badelt S, Flamm C, Hofacker IL. 2016. Computational design of a circular RNA with prionlike behavior. Artif Life 22:172–184. doi:10.1162/ARTL_a_00197.CrossRefGoogle Scholar
29.↵Ferré-D’Amaré AR, Scott WG. 2010. Small self-cleaving ribozymes. Cold Spring Harb Perspect Biol 2:a003574. doi:10.1101/cshperspect.a003574.Abstract/FREE Full TextGoogle Scholar
30.↵Ramesh A, Winkler WC. 2014. Metabolite-binding ribozymes. Biochim Biophys Acta 1839:989–994. doi:10.1016/j.bbagrm.2014.04.015.CrossRefGoogle Scholar
31.↵Ferré-D’Amaré AR. 2011. Use of a coenzyme by the glmS ribozyme-riboswitch suggests primordial expansion of RNA chemistry by small molecules. Philos Trans R Soc Lond B Biol Sci 366:2942–2948. doi:10.1098/rstb.2011.0131.CrossRefPubMedGoogle Scholar
32.↵Tinsley RA, Furchak JRW, Walter NG. 2007. Trans-acting glmS catalytic riboswitch: locked and loaded. RNA 13:468–477. doi:10.1261/rna.341807.Abstract/FREE Full TextGoogle Scholar
33.↵Lau MWL, Ferré-D’Amaré AR. 2013. An in vitro evolved glmS ribozyme has the wild-type fold but loses coenzyme dependence. Nat Chem Biol 9:805–810. doi:10.1038/nchembio.1360.CrossRefGoogle Scholar
34.↵Diener TO. 2003. Discovering viroids—a personal perspective. Nat Rev Microbiol 1:75–80. doi:10.1038/nrmicro736.CrossRefPubMedWeb of ScienceGoogle Scholar
35.↵Elena SF, Dopazo J, Flores R, Diener TO, Moya A. 1991. Phylogeny of viroids, viroidlike satellite RNAs, and the viroidlike domain of hepatitis delta virus RNA. Proc Natl Acad Sci U S A 88:5631–5634. doi:10.1073/pnas.88.13.5631.Abstract/FREE Full TextGoogle Scholar
36.↵Flores R, Grubb D, Elleuch A, Nohales MÁ, Delgado S, Gago S. 2011. Rolling-circle replication of viroids, viroid-like satellite RNAs and hepatitis delta virus: variations on a theme. RNA Biol 8:200–‐206. doi:10.4161/rna.8.2.14238.CrossRefPubMedWeb of ScienceGoogle Scholar
37.↵Daròs JA, Flores R. 1995. Identification of a retroviroid-like element from plants. Proc Natl Acad Sci U S A 92:6856–6860. doi:10.1073/pnas.92.15.6856.Abstract/FREE Full TextGoogle Scholar
38.↵Vera A, Daròs JA, Flores R, Hernández C. 2000. The DNA of a plant retroviroid-like element is fused to different sites in the genome of a plant pararetrovirus and shows multiple forms with sequence deletions. J Virol 74:10390–10400. doi:10.1128/jvi.74.22.10390-10400.2000.Abstract/FREE Full TextGoogle Scholar
39.↵Hegedus K, Dallmann G, Balázs E. 2004. The DNA form of a retroviroid-like element is involved in recombination events with itself and with the plant genome. Virology 325:277–286. doi:10.1016/j.virol.2004.04.035.CrossRefPubMedWeb of ScienceGoogle Scholar
40.↵Hammann C, Luptak A, Perreault J, de la Peña M. 2012. The ubiquitous hammerhead ribozyme. RNA 18:871–885. doi:10.1261/rna.031401.111.Abstract/FREE Full TextGoogle Scholar
41.↵Bussière F, Ledû S, Girard M, Héroux M, Perreault J-P, Matton DP. 2003. Development of an efficient cis-trans-cis ribozyme cassette to inactivate plant genes: cis-trans-cis self-cleaving ribozyme cassette. Plant Biotechnol J 1:423–435. doi:10.1046/j.1467-7652.2003.00039.x.CrossRefPubMedGoogle Scholar
42.↵Webb C-HT, Lupták A. 2018. Kinetic parameters of trans scission by extended HDV-like ribozymes and the prospect for the discovery of genomic trans-cleaving RNAs. Biochemistry 57:1440–1450. doi:10.1021/acs.biochem.7b00789.CrossRefGoogle Scholar
43.↵Huang X, Zhao Y, Pu Q, Liu G, Peng Y, Wang F, Chen G, Sun M, Du F, Dong J, Cui X, Tang Z, Mo X. 2019. Intracellular selection of trans-cleaving hammerhead ribozymes. Nucleic Acids Res 47:2514–2522. doi:10.1093/nar/gkz018.CrossRefGoogle Scholar
44.↵Czech B, Hannon GJ. 2016. One loop to rule them all: the ping-pong cycle and piRNA-guided silencing. Trends Biochem Sci 41:324–337. doi:10.1016/j.tibs.2015.12.008.CrossRefPubMedGoogle Scholar
45.↵Czech B, Munafò M, Ciabrelli F, Eastwood EL, Fabry MH, Kneuss E, Hannon GJ. 2018. piRNA-guided genome defense: from biogenesis to silencing. Annu Rev Genet 52:131–157. doi:10.1146/annurev-genet-120417-031441.CrossRefGoogle Scholar
46.↵Rechavi O, Lev I. 2017. Principles of transgenerational small RNA inheritance in Caenorhabditis elegans. Curr Biol 27:R720–R730. doi:10.1016/j.cub.2017.05.043.CrossRefGoogle Scholar
47.↵Battaglia E. 2005. Prion, protean, proteo-conformer: a terminological analysis. Rend Fis Acc Lincei 16:5–17. doi:10.1007/BF02904737.CrossRefGoogle Scholar
48.↵de la Peña M. 2018. Circular RNAs biogenesis in eukaryotes through self-cleaving hammerhead ribozymes, p 53–63. In Xiao J (ed), Circular RNAs. Springer Singapore, Singapore.Google Scholar
There is enough scientific basis to be worried by these global experimentation of health people by injecting them with mRNA components which were never studies in man before…
In the independent scientific community I am not alone at all and we share the great concern that we will see a tremendous increase of auto-immune diseases caused by prions and prion-like structures.
Pingback: Chloroquine is a potent inhibitor of SARS coronavirus Robert Gorter, MD, PhDRobert Gorter, MD, PhD
Pingback: COVID-19 RNA Based Vaccines and the Risk of Prion Disease Robert Gorter, MD, PhDRobert Gorter, MD, PhD
Very interesting paper, but there seems to be some missing elements.
Unfortunately it seems that some illustrations are missing. For instance, the following text excerpt indicates that this is the case: “Protein prions and RNAs that may behave like prions
In all panels, the dark blue lines and shapes indicate the benign forms of proteins or RNAs, red lines and shapes indicate the prion forms, solid arrowed lines indicate the direction of the reaction, and broken arrowed lines indicate the autocatalytic cleavages (gray for the relatively inefficient reactions and black for the more efficient ones).”
No such illustration can be found here at all. Is it possible to correct this or can you direct me to a place where these illustrations can be found?